

For targeted medicines, the current processes of regulatory evaluation, decision frameworks and delivery systems are limiting the implementation of new, better therapies. A healthcare ecosystem designed to enable innovation must also support R&D that is cost effective for all stakeholders and make-good on the promise that innovative medicines can effectively deliver the right prevention and treatment to the right patient at the right time.
By Duane Schulthess, 20th June, 2013
There is much room for optimism regarding the next generation of medical therapies. From the discovery of HER2 over-expression in the personalised treatment of breast cancer twenty years ago, therapeutic innovation is continuing to drive advances in the understanding of the genetic mechanisms of diseases. A recent study published in Nature and funded by Cancer Research UK has identified the existence of 10 distinct types of breast cancer, each with its own unique molecular structure, thus providing ever greater possibilities for the discovery and advancement of cures. And these innovation breakthroughs are not limited to oncology. A recent study published in The Lancet received international coverage, highlighting a stem-cell technique developed at Gothenburg University where living cells were removed from a vein of a deceased donor and replaced with the patient’s own before implantation. There was no rejection profile or need to suppress the immune system, and the blood flow between the intestines and the liver of the 10 year old patient was restored. [2] Prof. Sumitran-Holgersson, a member of the stem-cell research team at Gothenburg University that developed the new procedure, stated in a recent interview, “Could the EU help me get this therapy to the rest of the world? How can I build the infrastructure and get the therapy out there?”[3] The answers to Prof. Sumitran-Holgersson’s questions are vital to the future success and development of innovative therapies in Europe. In February of 2012, Forbes Magazine ran an article calculating that the full cost to a pharmaceutical company of launching a new therapy was $4 billion USD[4]. These exponentially increasing costs are partially driven by a regulatory system that does not respond well to the segmented and smaller populations of current science-led medical innovations, ultimately stopping many new ground-breaking medicines from reaching the patients who need them. A case in point is Belgium’s Tigenix, a spin-out from KU Leuven which had the first stem-cell therapy approved for sale by the European Medicines Agency (EMA) in 2009. Their unique procedure is a substitute for common invasive knee replacement surgery, instead using a patient’s stem cells to re-grow a patch of their own cartilage. While Tigenix has EMA approval for sale, gaining reimbursement in Europe continues to move slowly as they only received authorization by the Spanish health authorities in March of this year – four years after EMA approval. And while Tigenix is a small independent biotech, large medial companies embracing the next generation of innovative medicines have also had major setbacks in securing reimbursement for new therapies. Pfizer’s new lung cancer drug targets the ALK gene-mutation for non-small cell lung cancer, a subtype impacting 4% of cases; roughly 9,000 per year in Europe and 45,000 worldwide[5]. These patients do not respond well, if at all, to chemo and radiation therapies and have extremely high mortality rates. However, clinical trial data was promising; the majority of participants received the drug in late metastatic stages of the disease where other treatments had failed, and showed 60% surviving nearly a year after starting treatment.[6] Patients’ groups followed the regulatory process closely, with grass-roots internet campaigns disseminating information regarding trial participation, drug reactions/complications, and approval status[7]. The drug was approved for sale by the FDA with a companion diagnostic in August of 2011[8], and received conditional marketing authorization from EMA in October of 2012[9]. Since then, the new drug has had a very difficult time in Europe. Despite the promising results of Metastatic stage trials, Germany’s Institute for Quality and Efficiency in Health Care recently ruled that it provided “no additional benefit” to patients.[10] As well, the UK’s National Institute of Clinical Excellence issued guidance that does not recommend it for reimbursement.[11] These examples serve to illustrate a much larger issue – new innovative drugs and therapies that are needed by patients face enormous barriers to implementation and adaption. They also outline why an alternative must be found that enables the earlier availability of safe and effective medicines in a framework that is financially viable for all stakeholders, as the current healthcare system’s existence is being challenged. The European Medicines Agency issued a press release in March of 2013 that highlights their willingness to seek new regulatory pathways. It states, “Medicines regulation today is characterised by the increasing complexity of applications for new medicines, such as nanomedicines or personalised medicines, and the drug-development environment as a whole…An innovative evaluation framework involving iterative phases of data gathering and regulatory evaluation is needed in order to align regulatory approval more closely with patients’ needs for timely access to innovative medicines. This also includes the ability to integrate in the decision-making process multiple data sources — not only industry studies but also data from real-world use, as well as the views of patients on the level of risk acceptable for a given medical benefit.”[12] The above changes are vital to address the current deficiencies of the healthcare system and their impact on stakeholders. FIGURE 1: The Current Regulatory Pathway for new therapies in Europe. The current regulatory framework requires 14 years or more for a new compound to move from the lab, through trials, and then receive reimbursement (Figure 1). In Europe, the negotiations for EMA approval and reimbursement are now taking nearly as long as the actual clinical trial process, adding years to the time it takes to recoup an ever increasing investment. This is coupled by the fact that a targeted therapy for a small population requires an enormous number of trial locations to meet the current regulatory requirements. A case in point, the Pfizer trial for non-small cell lung cancer mentioned above required a staggering 261 international trial sites.[13] For patients and healthcare professionals, a regulatory environment should be created that supports the patient’s need for timely access to effective innovative medicines. For industry members, improved pathways would support innovation, increase effectiveness, and reduce R&D costs promoting investment by the European pharmaceutical sector. For payers, recent peer reviewed research shows that analysing the cost of treatment alone may overlook the gains in efficiency that a new targeted therapy can provide in other parts of the healthcare value chain.[14] In order for payers to be aware of such efficiencies, new pathways will require payers to be part of the entire evaluation process before HTA approval. It is promising that both EMA and FDA are currently participating in two key test-beds that seek to quantify the total net benefit of improved regulatory procedures. One of these is the NEW Drug Development ParaDIGmS (NEWDIGS) programme at MIT, another is the Centre for the Advancement of Sustainable Medical Innovation(CASMI), a research partnership of University College London and the University of Oxford, recently launched at the Wellcome Trust.([15])([16]) Ultimately, a revised regulatory framework to promote innovation in healthcare would support the translation of scientific breakthroughs from the lab to patients, increasing innovation in the life sciences and creating benefits to society. Without improvement, the barriers to providing the rapid deployment of new, and in particular, stratified therapies will continue to mount, eventually putting the entire healthcare ecosystem at risk for all of us. Duane Schulthess is the Managing Director of Vital Transformation, a consultancy focused on quantifying the impact of new technologies in the health-care sector. He is a member of the Advisory Board of Health Policy and Technology, and his recent study with Dr. Daniel Gassull, Dr. Stuart Suttie, and Prof. Graeme Houston on the impact of whole body MRA in diagnosing CVD will be published in the December 2013 edition. This editorial taken from the 2013 annual report of the European Federation of Pharmaceutical Industries and Associations and is reprinted with their permission. [1] C. Curtis, et al, “The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups”, Nature; 486, 346–352 (21 June 2012) [2] M. Olausson, Suchitra Sumitran-Holgersson, et al, “Transplantation of an allogeneic vein bioengineered with autologous stem cells: a proof-of-concept study”; The Lancet, Volume 380, Issue 9838, Pages 230 – 237, 21 July 2012, http://www.thelancet.com/journals/lancet/article/PIIS0140-6736%2812%2960633-3/abstract [3] N. Moran, “Experts debate the future of stem cells”; Science|Business, Pg. 20, January 2013, http://www.sciencebusiness.net/OurReports/ReportDetail.aspx?ReportId=39 [4] M. Herper, “The Truly Staggering Cost Of Inventing New Drugs”, Forbes, February 10, 2012; http://www.forbes.com/sites/matthewherper/2012/02/10/the-truly-staggering-cost-of-inventing-new-drugs/ [5] R. Wilson, P. Loftus, “Advances Come in War on Cancer”, Wall Street Journal, June 7, 2010; http://online.wsj.com/article/SB10001424052748704002104575291103764336126.html?mod=WSJ_WSJ_US_News_3 [6] FDA Approval for Crizotinib, National Cancer Institute, NIH; http://www.cancer.gov/cancertopics/druginfo/fda-crizotinib; accessed May 13, 2013 [7] Lung Cancer Survivors Support Community; http://www.inspire.com/groups/lung-cancer-survivors/search/?query=Crizotinib&submit.x=37&submit.y=10, accessed May 9, 2013 [8] FDA Press Release, “FDA approves Xalkori with companion diagnostic for a type of late-stage lung cancer” http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm269856.htm, accessed May 12, 2013[9]Europan Medicines Agency, Regulatory Overview Xalkoiri (crizotinib); http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002489/human_med_001592.jsp&mid=WC0b01ac058001d124; accessed May 13, 2013 [10] T. Staton, “German Cost Agency Spurns Xalkori…”, February 21, 2013, http://www.fiercepharma.com/story/german-cost-agency-spurns-xalkori-komboglyze/2013-02-21; accessed May 13, 2013 [11] B. Hirshler, “Pfizer lung cancer pill rejected by NICE”; Reuters, Wed Mar 27, 2013, http://uk.reuters.com/article/2013/03/27/uk-pfizer-britain-xalkori-idUKBRE92Q00L20130327; accessed May 13, 2013 [12]Press release, “European Medicines Agency’s Management Board welcomes new civil-society members” ; accessed May 12, 2013 [13] http://clinicaltrials.gov/ct2/show/study/NCT01154140?show_locs=Y#locn; accessed June 20, 2013 [14] W. Van Dyck , et al,” Unlocking the value of personalised healthcare in Europe—breast cancer stratification http://www.healthpolicyandtechnology.org/article/S2211-8837%2812%2900044-5/; accessed May 12, 2013 [15] MIT center for Biomdical Innovation, http://cbi.mit.edu/research-overview/newdigs/, accessed May 13, 2013 [16] CASMI, http://casmi.org.uk/, accessed May 13, 2013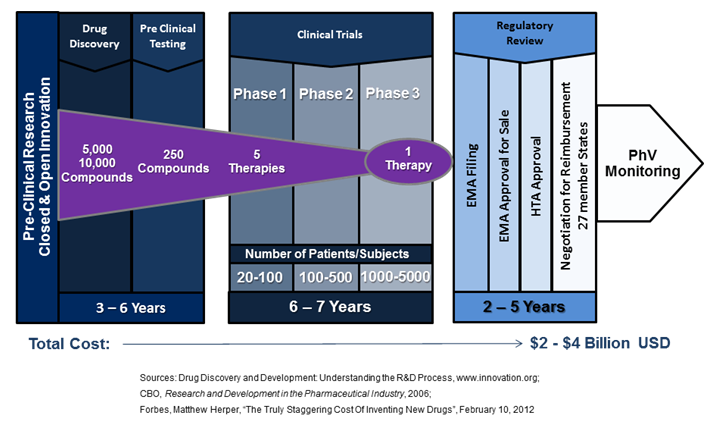




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}